
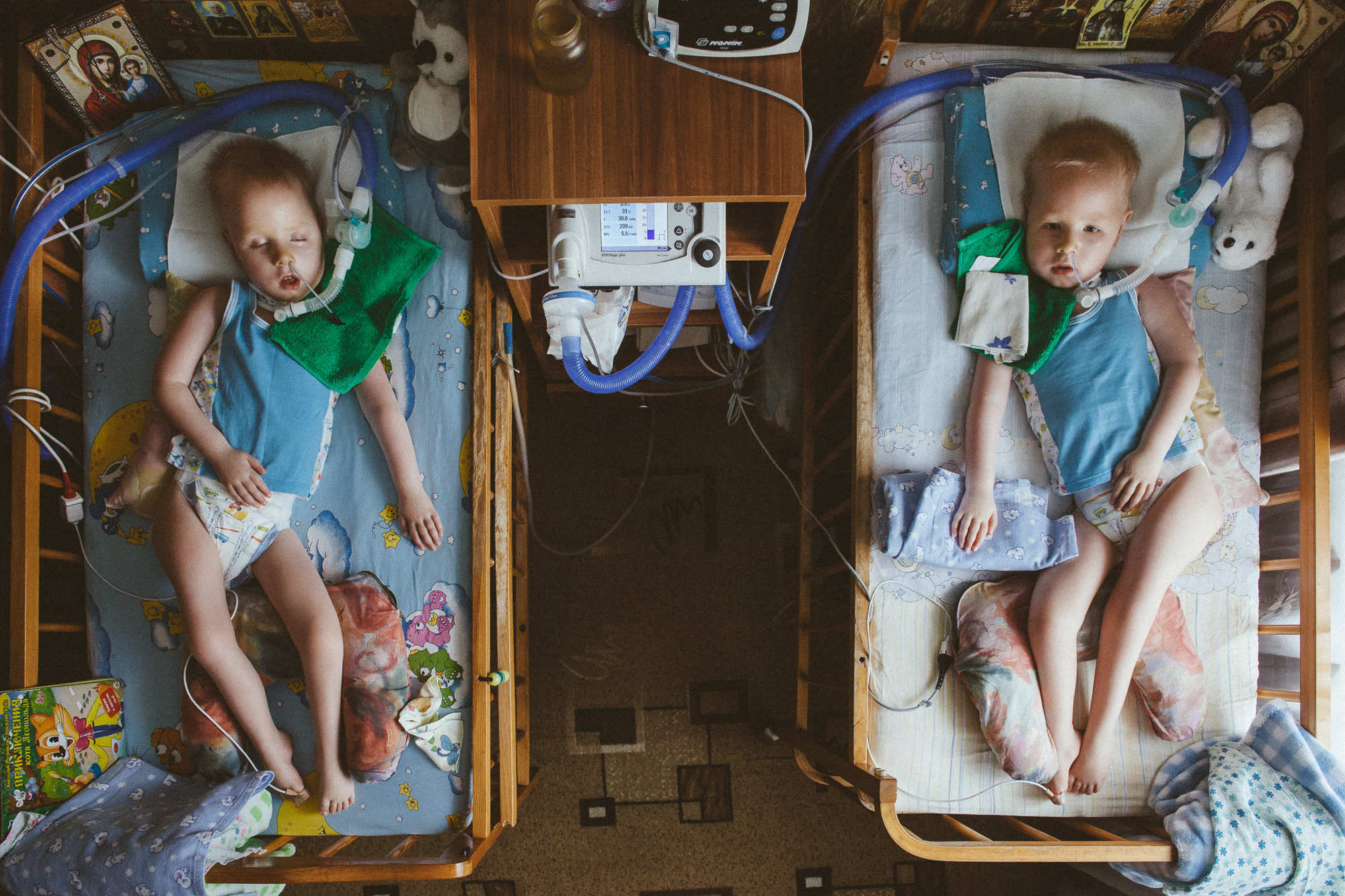
Восьмилетний Федя Минько из Минска еще год назад бегал и болтал без умолку. Теперь он больше не говорит, почти потерял слух и ходит с трудом. Лекарство, которое могло бы помочь, вовремя не пришло. Хотя Минздрав Беларуси обещал выдавать его бесплатно. У Феди и его брата Матвея мукополисахаридоз. Людей с таким диагнозом в Беларуси немного. Известно про 18 детей и 19 взрослых. При этой болезни в органах и тканях откладывается вещество, которое поражает со временем весь организм. Это болезненно и неизлечимо. В России, Казахстане, Польше таким людям бесплатно выдают лекарство, которое, считается, улучшает состояние и замедляет развитие болезни. Планировалось, что и у нас будет терапия для пациентов с редкими генетическими заболеваниями. Заложили в проект бюджета 8,8 миллиона долларов — лекарства дорогие. Но ни Федя, ни Матвей препарат пока не получили. Минздрав изучает международный опыт и пытается договориться о цене с поставщиком. Уже второй год. Очевидно, денег в бюджете не хватает, сил местных фондов и организаций — тоже. В Беларуси должна появиться эффективная система помощи больным с редкими генетическими заболеваниями. Нужна помощь международных организаций, бизнеса и каждого из нас.
— В полтора года у Федьки заболели уши, начались проблемы с суставами, изменилась внешность, — говорит его мама Анна Минько. — В два года мы попали к ортопеду. Он по характерной форме головы предположил, что это что-то генетическое и отправил к специалисту. Мне тогда сказали, что такие дети, как мой сын, мало живут. Я была в таком шоке! А теперь просто ужасно знать, что можно было избежать этих сильных осложнений — было бы лекарство.

Анна, кажется, могла бы командовать полком, но всю энергию направляет на то, чтобы бороться за своих детей по всем фронтам. Специальные занятия по мозговой стимуляции, ЛФК, пиявки, иппотерапия, бассейн, диета, реабилитация два раза в год. Девушка показывает деревянный вертикализатор, который смастерил папа подруги — с помощью этой конструкции можно закрепить ребенка в правильном положении.
Мама Феди и Матвея купила золотую рыбку и загадала желание, чтобы Минздрав не передумал.
У брата Феди трехлетнего Матвея тот же диагноз — мукополисахаридоз. Это группа редких наследственных заболеваний. Из-за нее во всех органах «оседают», как накипь на чайнике, вещества мукополисахариды. На клапанах сердца, стенках сосудов, в глазах, печени, костях. И разрушают их. Болезнь нельзя вылечить. Но несколько лет назад появились препараты, способные остановить или замедлить ее развитие. Ампула лекарства стоит 2500 долларов. Одному только Федору нужно по четыре таких в неделю. В России, Казахстане, Польше средство предоставляет государство. У нас — пока нет.
Ведутся переговоры
В 2015 году Тамара Матиевич, председатель Белорусской организации больных мукополисахаридозом и другими редкими генетическими заболеваниями, обратилась в Администрацию Президента. Вопрос: будут ли власти обеспечивать людей с генетическими болезнями специальными лекарствами. Такое лечение называют «ферментозаместительной терапией». Пришел ответ: «Начиная с 2016 года министерство (здравоохранения — примечание редакции) обещает выполнить взятые на себя обязательства по обеспечению ферментозаместительной терапией пациентов в возрасте до 18 лет. В проекте бюджета предусмотрены финансовые средства в размере 8,8 миллиона долларов. В дальнейшем будет прорабатываться вопрос обеспечения всех пациентов, нуждающихся в проведении терапии».

Мама Феди и Матвея, когда услышала эту новость, купила золотую рыбку и загадала желание, чтобы Минздрав не передумал. Прошло почти два года. Ни Федя, ни Матвей лекарств ни разу не увидели. Федя, который еще год назад бегал и болтал, больше не говорит, с трудом ходит и почти потерял слух. С терапией у него есть шанс на долгую жизнь. Иначе — прогноз плохой или очень плохой. Потеря слуха, речи, подвижности, сердечная недостаточность, жизнь на аппарате искусственной вентиляции легких, кормление из трубочки. Сколько еще придется ждать помощи? Почему не выполняется то, что обещано?
— После обещаний Минздрава я поверила в лучшее и немного расслабилась, — с досадой вспоминает Анна. — А надо было продолжать писать, звонить и добиваться. В декабре прошлого года я звонила на горячую линию и спрашивала, будут ли лечить и куда пошли выделенные деньги. Мне сказали, что деньги переходят на следующий год.

В июне 2017 года «Имена» написали запрос в Минздрав. Спрашивали: будет ли бесплатное лекарство для детей и взрослых, больных мукополисахаридозом, получает ли вообще кто-нибудь ферментозаместитетельную терапию, сколько средств выделено на закупки таких препаратов, куда пошли обещанные 8,8 миллиона долларов? Минздрав ответил, что лекарства стоят дорого, эффективность не во всех случаях доказана, но внедрять ферментозместительную терапию все-таки начнут. Однако поэтапно.
«Надо поскорее начать лечение, чтобы не растерять навыки».
Например, сейчас лечат семь детей с болезнью Гоше за счет бюджета и десять взрослых, девять из них получают лекарство по гуманитарной помощи. Лечат еще одного ребенка с болезнью Помпе (тоже гуманитарная помощь). Минздрав обещает выдавать препарат детям больным мукополисахаридозом I типа, но только до трансплантации костного мозга — такая операция может помочь им.
Но есть проблема. У большинства белорусов с мукополисахардиозом — другой тип. Например, у Феди и Матвея — второй. Минздрав пишет, что вопрос о лекарствах для таких людей будет решаться только в 3-4 квартале 2017 года: изучают международный опыт, проводят переговоры о снижении цены. «Имена» написали второй запрос, чтобы спустя время узнать, как проходит изучение международного опыта, какие результаты, с кем ведутся переговоры о цене и какой же все-таки бюджет был выделен? Ответа нет.

Пока родители ждут терапию, Федя меняется. Раньше он был эмоциональным и жизнерадостным, старался заботиться о младшем брате. Сейчас не улыбается, замыкается в себе. Теперь Матвей за старшего.
— У Матюхи сейчас такой важный возраст, — говорит Анна. — Надо поскорее начать лечение, чтобы не растерять навыки. Он еще не говорит, а уже пора бы. Да и Федя в таких муках, на самом деле, у него всё болит. Мы очень ждем, что всё же министерство пойдет навстречу таким, как мы.
Начали лечить, а потом — бросили
Девять лет назад в Беларуси пробовали лечить мукополисахаридоз. Государство выделило лекарство двум пациенткам — Татьяне Матиевич из Могилева и минчанке Яне Карелиной (имя изменено по просьбе героини). Полгода девушки получали американский препарат. Им становилось лучше, но потом помогать перестали. Спустя три месяца, когда стали заметны улучшения, Минздрав признал лекарство, одобренное во всем мире, от США до Казахстана, неэффективным. Но два года назад ситуация поменялась — Минздрав пообещал закупить средства.
Дальше разговоров дело пока не идет.
Больные получают письма из министерства о том, что идут переговоры о закупке.

Сегодня Таня прикована к инвалидной коляске и испытывает постоянные боли. Поскольку у девушки проблемы с голосом, говорит за нее мама — Тамара Матиевич:
— Все больные следят за исследованиями. Мы узнали о том, что наконец создают препарат, когда в Америке еще только проводили эксперименты на мышах. Хотели поучаствовать в испытаниях на людях, но не прошли. 14 лет назад мы начали ходить в Минздрав, рассказывать, что разрабатывается лекарство и что нужно планировать его закупать. Мы были первыми в Беларуси. И были активными. Настолько, что в конце 2008 года нас стали лечить. Тогдашний министр здравоохранения Василий Жарко пообещал, что раз уже начали, то не перестанут. Бросать нельзя. Известно, что от этого станет еще хуже.
«Знать, что есть лекарство, помогающее жить нормально, без боли, и не иметь возможности его получать — это ужасно»
До ферментотерапии у Тани всё болело, она не ходила. Несмотря на все трудности, могилевчанка сумела закончить школу с золотой медалью и получить диплом психолога. Благодаря лечению девушка встала с инвалидной коляски, смогла передвигаться по квартире, улучшилось состояние внутренних органов, стало легче дышать. Жизнь была полна надежд. Но несмотря на обещания, лекарство резко перестали давать. Пришло письмо о том, что ученый медицинский совет вынес решение о недоказательности клинической эффективности препарата.
— После того, как нас бросили, пришлось срочно ложиться на операцию и менять клапаны сердца. Таня еле выжила, — говорит мама Тамара.

Вторая пациентка, получавшая лечение, Яна, показывает документ и с трудом сдерживает эмоции:
— Во время лечения врачи поздравляли нас с успехами. А потом препарат признали неэффективным. Нам сказали, что это всё плацебо. Мы даже в суд обращались, чтобы лечение продолжили, но ничего не вышло.
Яна, экономист по профессии, привыкла ко всему подходить основательно. Она взяла все результаты обследований и свела в одну таблицу, в которой отчетливо видно, что улучшения — это не плод ее воображения, а реальный факт. Можно проследить, как снижалось содержание мукополисахаридов в анализах, улучшились показатели сердца, печени, зрение и так далее.
— Я с девяти лет жила с болью, потому что у меня разрушались суставы, — говорит Яна. — С 21-го года я принимала серьезные обезболивающие, в том числе такие, которые дают онкобольным. Невозможно было даже лежать. Я сейчас вспоминаю тот кошмар и удивляюсь, как удалось пережить. Много лет спала по полчаса, часами не могла заснуть. После первых капельниц препарата я смогла проспать всю ночь. А однажды, когда проходила промежуточное обследование, зашла на костылях в кабинет офтальмолога, поставила их у стенки, а потом забыла и ушла. Уже на другом этаже сообразила, что иду сама! Когда нас прекратили лечить, у меня ноги «посыпались» в течение месяца. Остальное продержалось на эффекте от приема лекарств еще несколько месяцев.
Больных мукополисахаридозом в мире мало. Многие друг друга знают и общаются несмотря на то, что живут в разных странах. Положение тех, кто живет не в Беларуси, заметно отличается:
— Моего года рождения в мире было шесть человек с таким мукополисахардиозом, как у меня, — рассказывает Яна. — У Дороти из Германии уже большой ребенок. Приятно смотреть на девчонок, которые построили семьи. Они получают лечение, и у них есть будущее. А у меня всегда были мысли: какое будущее без лекарства? Вот родится сын или дочь, а с кем он или она останется, если я умру? Думаю об этом до сих пор. Трудно планировать что-то глобальное. Мы с Таней на себе прочувствовали действие лечения ферментозаместительной терапией, помним эффект и все улучшения. Было очень тяжело это потерять. Знать, что есть лекарство, помогающее жить нормально, без боли, и не иметь возможности его получать — это ужасно.

«Надо посмотреть, что делают в других странах, откуда берут средства на лечение»
Семьи верят, что ситуация изменится к лучшему. Ведь во многих других странах к закупке таких лекарств тоже пришли не сразу. Например, в России всё началось со Снежаны Митиной, мамы мальчика с мукополисахаридозом второго типа и президента благотворительной организации «Хантер-синдром». Она умудрилась собрать деньги, которых хватило бы на несколько месяцев ферментозаместительной терапии в Германии. Поскольку прерывать лечение нельзя, и была цель привлечь государство к проблеме, женщина едва ли горы не сносила, чтобы добиться своей цели. И ее сына первого начали лечить в России за счет средств государства. Яна Карелина вспоминает, как первый раз побывала у Снежаны в гостях и увидела ее семилетнего Павлика:
— Не ходил, сидел, обложенный подушками, чтобы было не так больно. Его кормили из шприца. Когда я приехала еще раз через год, мальчик уже гулял по квартире. Сегодня в России лечат всех: и детей, и взрослых.
В Беларуси люди с мукополисахаридозом лишены возможности получать помощь — регулярную и в полном объеме. Каких-то лекарств нет вовсе и непонятно, когда будут. К другим «присматриваются», изучают. Третьи — могут назначить, а потом внезапно отменить. Ситуация меняется — и снова начинаются переговоры, переписка, бесконечное ожидание.
Какое может быть решение?
За 40 лет, что в Беларуси проводится диагностика мукополисахаридозов, через специалистов прошло 150 человек с этими заболеваниями. В живых осталось 37. Генетик Нина Гусина, которая заведует клинико-диагностической генетической лабораторией РНПЦ «Мать и дитя» уже много лет, говорит:

— Препараты должны быть материально доступны. Сейчас цена велика. Но и больных мало. Понятно, что деньги не бесконечны и, наверное, на всех не хватает. Но всё-таки подумать об этих пациентах надо. Посмотреть, что делают в других странах, откуда берут средства на их лечение. Редкие болезни — это мерило гуманности общества. Почему мы должны серьезно относиться только к тем заболеваниям, которые встречаются тысячами? Эти единичные слабые ручки, протянутые к нам, точно так же достойны нашего внимания и помощи, — говорит генетик.
Чтобы бороться за право на жизненно необходимую терапию не только для своей дочери, но и для других, Тамара Матиевич, мама Татьяны, которая некоторое время получала лекарства, создала Белорусскую организацию больных мукополисахаридозом и другими редкими генетическими заболеваниями. В одном из наших материалов она объясняла, что стране нужен специализированный центр для людей с редкими генетическими заболеваниями, где будут отдельные палаты для пациентов, будет собран консилиум — штат профессионалов. Врачи, в свою очередь, могли бы в этом центре подбирать самые последние новинки лекарств, поддерживающих нормальное состояние человека.
История Феди и Матвея, Яны и Тани — история неотвеченных писем — лишь часть большой проблемы. Больных мукополисахаридозом — всего 37 человек. Больных генетическиим заболеваниями — сотни. Каждому из них говорят в чиновничьих кабинетах и иногда в фондах примерно так: «Это редкая болезнь, на все редкие болезни денег не набраться; есть проблемы, которые касаются гораздо большего количества людей, и их тоже надо решать». Мы не раз писали про родителей, которые выбивают деньги на аппараты искусственной вентиляции легких, проводят жизнь в сборах на расходники и лекарства, ввозят их полулегально из-за границы. Кто-то обращается в фонды, которые физически не могут помочь всем. Кто-то годами переписывается с чиновниками. В Беларуси пока нет эффективной системы помощи больным с редкими генетическими заболеваниями. И ее надо создавать вместе.
Вспомним героев, о которых мы раньше писали. Это люди с разными генетическими заболеваниями. Все они постоянно нуждаются в дорогом лечении и поддержке.


Очевидно: бюджетных денег на медицинское оборудование и дорогие лекарства не хватает. И люди годами остаются без помощи. Местным фондам и объединениям — а их сегодня тоже немного — без поддержки проблему целиком не решить. Нужна платформа/центр, который объединит специалистов — медиков, чиновников, бизнесменов, фандрайзеров, международные организации, которые хотят и могут помогать, а также самих больных и их родителей. «Имена» готовы поддержать такой центр. Пишите на [email protected].
Как вы можете помочь Феде и Матвею Минько:
Перевести деньги на благотворительный счет Анны Александровны Минько. Благотворительные счета открыты в ОАО «АСБ Беларусбанк», операционная служба филиала 527 — город Минск, ул. Воронянского, дом 7а. УНП 100783330. БИК AKBBBY21527.
— белорусские рубли: благотворительный счет № BY63 AKBB 3134 0000 0325 4542 0527
— российские рубли: благотворительный счет № BY18 AKBB 3134 0000 0325 7542 0527
— доллары: благотворительный счет № BY48 AKBB 3134 0000 0325 5542 0527
— евро: благотворительный счет № BY33 AKBB 3134 0000 0325 6542 0527.
«Имена» работают на деньги читателей. Вы оформляете подписку на 3, 5, 10 рублей в месяц или делаете разовый платеж, а мы находим новые истории и помогаем еще большему количеству людей. Выберите удобный способ перевода — здесь. «Имена» — для читателей, читатели — для «Имен»!